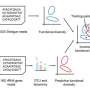
New sequencing technique established for low-biomass, degraded or contaminated microbiome samples
 to identify all candidate species in a 2bRAD-M sample. Next, to accurately estimate the abundance of identified species, we increased the number of taxa-specific 2bRAD markers for each candidate species by reconstructing a reduced 2bRAD marker database (sample-specific 2b-Tag-DB) which contains more 2bRAD markers specific to each candidate species than those in the default 2b-Tag-DB. All the 2bRAD sequences were then remapped to this sample-specific 2b-Tag-DB for abundance estimation of candidate species. In principle, the relative abundance of a given species was calculated as the read coverage of all species-specific 2bRAD markers. Credit: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02576-9")
A research team led by the Qingdao Institute of Bioenergy and Bioprocess Technology (QIBEBT), Chinese Academy of Sciences (CAS), has developed a low-cost metagenome sequencing technique. It enables high-resolution revelation of the full 'landscape' of organisms in a microbiome, even from low-biomass, degraded or contaminated samples.
Their study was published in Genome Biology on Jan. 26.
The 'microbiome' is the community of bacteria, fungi, protists and viruses that lives in and on humans, or indeed in any habitat. To find out which microbes is part of a given microbiome, researchers have to carry out metagenomic sequencing, or the sequencing of genetic material from multiple organisms at the same time.
However, even state-of-the-art metagenomic sequencing still has many severe limitations. There are two main categories of the process: amplicon sequencing and whole metagenome sequencing (WMS).
The amplicon sequencing is cheap. Unfortunately, it only produces information about the members of the microbiome at the taxonomic level of genus. Species or strain level taxonomic information is difficult to tease out. Amplicon sequencing also suffers from only being able to identify bacteria or archaea or fungi or viruses, rather than identify them all at once.
WMS can capture whole DNA sequences of everything in the sample鈥攂acteria, archaea, fungi and viruses鈥攁nd at a higher taxonomic resolution. This gives much more information, but it is much more costly. WMS also requires a large amount of high-quality DNA.
"What we need is a low-cost sequencing method that can enable accurate, species-resolution identification of all microbes at the same time, and can handle low biomass samples," said Sun Zheng, first author of the paper and a researcher with the Single-Cell Center at QIBEBT.
The researchers developed what they call "2bRAD sequencing for Microbiome" method, or 2bRAD-M for short. This makes use of restriction site-associated DNA sequencing (RADseq) that utilizes Type IIB restriction enzymes (proteins that cleave DNA at specific sites along the molecule) to digest genomic DNA from stool, skin or environment-surface samples into fragments of 25-33 base pairs (pairs of nucleotides).
The 2bRAD-M technique sequences only these digested fragments鈥攐r about just one percent of the metagenome鈥攁nd can produce profiles across bacteria, archaea, and fungi, and down to the level of species, not just genus.
"By needing only a single picogram (a thousandth of a nanogram) of total DNA, the process can cheaply and accurately generate high-resolution profiles even for hard-to-sequence samples where there has been high host DNA contamination, or the DNA is severely fragmented or otherwise degraded," said Huang Shi, co-first author of the paper.
One of the most intriguing discovery over the past years has been that microbes are implicated in cancer development. Using 2bRAD-M, the researchers showed that the microbiome profiles from cervical cancer tissues can be sensitively and reliably revealed from formalin fixed paraffin embedded (FFPE) samples, which are typically of low amount, degraded or contaminated by human DNA. Such profiles can potentially be employed for the early diagnosis of cervical cancers.
"In the future, we hope to further develop the 2bRAD-M technique and explore its various clinical applications," said senior author Xu Jian, Director of Single-Cell Center at QIBEBT. "Ultimately, we hope that 2bRAD-M would become a cost-efficient, popular tool for microbiome research and conquer various challenging tasks that beyond the reach of current techniques," added Prof. Wang Shi from Ocean University of China, another senior author of the study.
More information: Zheng Sun et al, Species-resolved sequencing of low-biomass or degraded microbiomes using 2bRAD-M, Genome Biology (2022). .
Journal information: Genome Biology
Provided by Chinese Academy of Sciences